Hemangioblastoma of the Cerebellum; von Hippel-Lindau Complex
Hemangioblastomas are histologically benign tumors that occur exclusively within the neuraxis, most commonly in the posterior fossa. (Rare exceptions to this rule include recent reports of hemangioblastomas in the peripheral nerves. They account for 1.5 to 2.5 percent of all intracranial tumors and for 7 to 12 percent of posterior fossa tumors. The term hemangioblastoma was originally suggested by Cushing and Bailey to describe these tumors, which were thought to arise from "vasoformative" cells (endothelial cells) of the central nervous system. This term stressed the neoplastic nature of the lesion and thereby served to distinguish it from the more common hemangiomas of the nervous system (which are not true tumors but hamartomas). Despite the objection that the term conveyed an erroneous impression of cellular components of primitive nature, implying a malignant potential, it has been widely accepted and is well established in the literature. It is preferred over such synonyms as hemangioma, capillary hemangioendotheliama, Lindau's cyst, Lindau's tumor, or angioreticuloma. Lindau's tumor specifically refers to the tumor of the cerebellum and von Hippel's tumor to hemangioblastoma of the retina. Lindau' s disease, or von Hippel-Lindau's (VHL) complex, designates a more diffuse inherited disorder characterized by multiple hemangioblastomas in the neuraxis associated with certain visceral manifestations. The minimal clinical criteria required to justify the diagnosis of VHL complex are either (I) more than one hemangioblastoma within the neuraxis (strongly implying multicentricity), (2) a solitary hemangioblastoma of the neuraxis associated with at least one visceral manifestation, or (3) any clinical manifestation of the VHL syndrome in a first-degree relative of an individual known to have von Hippel-Lindau's syndrome. More than 30 manifestations of VHL syndrome have been described; the most frequently occurring lesions are outlined in Table 1. Recent advances in molecular genetics have resulted in the identification of the VHL gene and the development of screening tests to permit diagnosis in presymptomatic individuals.
von Hippel's classic paper appeared in 1904. In it he presented two patients with a retinal mass associated with an enormously dilated artery and vein and with retinal detachment and exudation. Unaware of Collins' work and lacking tissue for diagnosis. he was unable to assign a cause to this unusual disease. Seven years later, from histologic examination of enucleated eyes from his patients described in previous reports and as a result of becoming aware of Collins' work, he concluded that the primary lesion in the retina was a hemangioblastoma. He thus publicized a new and fascinating clinical entity, which soon bore his name as an eponym. After the death of one of von Hippel's patients, Brandt published the autopsy results. In the central nervous system tumors were found in the cerebellum, in the base of the brain near the petrous bone, and in the cauda equina. In the kidneys there were cysts and tumors. The pancreas was studded with cysts. The bladder contained papillomas. and the epididymes bore cysts. Brandt also described one other patient with angiomatosis retinae, who had died of brain tumor.
A search of the literature revealed that in at least 10 percent of all cases of angiomatosis retinae. The stigmata of central nervous system neoplasms were also present. Of particular interest was Seidel's report of cerebellar cyst associated with retinal hemangioblastoma in two brothers. Before the onset of cerebellar dysfunction, these men had been earning their living as tightrope walkers.
Thus the time was ripe for Arvid Lindau. a young Swedish pathologist whose monograph in 1926 brought together into one coherent entity the retinal, cerebellar and visceral components of this disease. Lindau's work had begun as an investigation of cerebellar cysts. Most of these were associated with a mural tumor and most of the mural tumors were angioblastic. Lindau was particularly impressed by the small size of these mural nodules and emphasized that they could be missed unless the cyst wall was meticulously dissected. He collected the data on 40 such cases, 24 from the literature and 16 of his own. He found that the hemangioblastomas were not confined to the cerebellum but were present in the medulla and spinal cord as well. The visceral lesions he encountered were pancreatic cysts, renal cysts, renal cell carcinomas, adrenal adenomas, hepatic adenomas, cavernomas of the liver and tumors of the epididymis.
Genetics
Hemangioblastomas may occur sporadically as isolated tumors of the cerebellum or may represent a familial disorder as part of the VHL complex. The latter disorder is transmitted as an autosomal dominant trait with varying penetrance and it may be passed on by affected or unaffected members of either sex. Great strides have been made recently in understanding the molecular genetics of VHL disease owing largely to the power of recombinant DNA technology. Using recombinant techniques, cytogenic analysis was performed on families known to carry the VHL gene. Early studies from several groups indicated that the VHL gene localized to the short arm of chromosome 3 (3p). Seizinger and colleagues observed that the VHL gene was linked with the homolog of the RAFI oncogene, which was known to map to a specific segment of 3p designated 3p25. This observation was particularly interesting because it had previously been observed that sporadic renal cell carcinomas were associated with loss of regions on chromosome 3p. Further, the proximal portion of the short arm of chromosome 3 had been implicated in a variety of other tumors, including adenocarcinoma of the lung. Rhabdomyosarcoma, ovarian carcinoma, mesothelioma and others. The most common breakpoints in these tumors clustered in region 3pll-21. Further cloning studies localized the gene to the 3p25-26 region. By closely examining the DNA from individuals affected with the VHL syndrome, areas of commonly deleted DNA were identified. As progressively smaller segments were identified, the deleted segments were termed nested constitutional deletions. Eventually, the smallest of the identified nested constitutional deletions was searched with known probes from a cDNA library for evolutionarily conserved sequences. Two sequences, designated g6 and g7 were identified. Patients with VHL syndrome were then screened for the presence of these sequences. Initial studies demonstrated that no g6 mutations were observed in a series of 120 VHL patients studied. By contrast, g7 transcripts were widely found in nearly all tissues in affected individuals. Later studies demonstrated that the g7 sequence is highly conserved across organisms, which suggests that g7 encodes a fundamental cellular function and is highly suggestive that g7 represents the gene for the VHL syndrome.
The precise function of the gene product of g7 remains unknown, yet several lines of evidence support its role as a tumor suppressor gene. A tumor suppressor gene is a gene whose normal role is to help uncouple or slow the replication process. Oncogenesis is thought to occur through either the action of an oncogene or the loss of action of a tumor suppressor gene. The theory of tumor suppressor gene function holds that tumors may arise by a one- or two-stage mutation process. In sporadically occurring tumors, a deletion must occur on both copies of the allele bearing the tumor suppressor gene before oncogenesis will be initiated. By contrast, in hereditary tumors, tumor formation is initiated by the loss of the single remaining allele (the other being absent from the time of conception). Clinically, this difference results in different age incidence curves for tumors arising in the two ways. Epidemiologic studies demonstrated that the age incidence curves for cerebellar hemangioblastoma in VHL are compatible with a single-mutation model, while curves for sporadic hemangioblastomas suggest a two-stage process. If the VHL gene is a tumor suppressor gene, it is atypical, because most tumor suppressor genes function recessively and the VHL syndrome shows dominant inheritance.
Clinical Features
The cerebellar or retinal tumors are usually the first to present clinically in patients with the VHL complex. Indeed, cerebellar hemangioblastoma is the manifestation of this disorder that produces the greatest morbidity and mortality. In several reported series, there is a slight male preponderance, with a male-to-female ratio of 1.3: 1. The average age at onset of symptoms in familial cases is 30 years, with a range of 3 to 83 years. Occurrence of symptoms in children less than 10 years old is distinctly rare. The peak incidence is in the third decade, with a second peak occurring in the fifth decade. The non-familial, or sporadic, cerebellar hemangioblastomas tend to present somewhat later with the mean age at initial presentation being 42 years. The average duration of symptoms is 13 months, with a range of 3 weeks to 7 years.
The symptoms and signs vary to some extent, depending on the precise location of the tumor in the posterior fossa. As a rule, symptoms and signs of increased intracranial pressure with varying expressions of cerebellar or brain stem deficits tend to dominate the picture. Headache is the most common symptom, being present in 95 percent of the cases. It is located in the suboccipital region and tends to be worse in the mornings, but it becomes relentlessly continuous in later stages. Patients with tumor located in the inferior vermis and tonsils, with chronic impaction of the tumor extending into the foramen magnum may have continuous occipital pain, neck stiffness, an intermittent shock-like sensation radiating into the occiput, and lapses of consciousness. Vomiting, the next most common symptom, may be due either to obstructive hydrocephalus from a vermian lesion or to irritation of the vagal nucleus from the tumor's origin in the vicinity of this nucleus. In the latter situation, vomiting may occur before any other neurological signs appear, leading to a mistaken diagnosis of upper gastrointestinal disorder. Vertigo tends to be a prominent symptom if the tumor is situated directly in the brain stem or in the middle or inferior cerebellar peduncle, with pressure on the vestibular nuclei. Gait disturbance and inability to maintain balance are generally expressions of cerebellar or brain stem hemangioblastoma. Papilledema was recorded in 20-30% of cases. Diplopia is usually due to sixth nerve paralysis from increased intracranial pressure. Ataxia in the extremities, dysmetria, and intention tremor are observed in cerebellar hemispheric lesions, whereas vermis lesions produce a broad-based gait and truncal ataxia. Nystagmus, especially with a vertical or rotatory component, signifies brain stem involvement. Occasionally in elderly patients, dementia may be the sole or major manifestation of the disease.
Spinal pain is the most common symptom in spinal hemangioblastoma and is a reliable indicator of the level of the tumor. The next most common symptoms of spinal hemangioblastomas are spastic quadriparesis, sensory changes, impaired micturition, and radicular neuralgia. Uncommonly, these lesions present with subarachnoid hemorrhage or acute paraparesis or quadriparesis
Pathology
Most hemangioblastomas occur in the posterior cranial fossa clustered around the fourth ventricle, generally in the vermis or hemisphere(s) of the cerebellum, medulla oblongata, or pons. Less commonly, they are found in the supratentorial compartment or in the spinal cord. Retinal hemangioblastomas occur in 6 percent of patients with cerebellar hemangioblastomas. Structurally, hemangioblastomas occurring at all locations are identical. In familial cases, tumors tend to be multiple. There is no satisfactory explanation for the preponderance of these tumors in the posterior fossa. Lindau suggested that a segment of the primitive choroid plexus becomes incorporated into the developing cerebellum in the 12th week of intrauterine life. Hemangioblastomas were thought to develop from these choroid remnants. This theory is especially attractive in light of the observation that one of the functions of the primordial choroid plexus is hematopoiesis, but it does not account for the occurrence of hemangioblastomas elsewhere in the neuraxis.
With rare exceptions, hemangioblastomas are benign tumors. Occasionally the tumor may spread along the subarachnoid space after a surgical procedure, but it remains histologically benign. Distant metastases have not been reported. Hemangioblastomas do not have a true capsule, but grossly the tumor margin is well circumscribed.. The tumors may be either solid or cystic. A higher proportion of cerebellar hemangioblastomas (70 percent) than of supratentorial or brain stem lesions (20 percent) are cystic. In cystic lesions, the solid component is a small nubbin at some point along the cyst wall, generally close to the pial surface, described commonly as a mural nodule. The cyst contains clear, golden-yellow, highly proteinaceous fluid which clots readily after aspiration. The inner surface of the cyst wall is smooth and is made up of glial cells and compressed cerebellar tissue; the tumor itself does not line the cyst wall. The cut surface of the solid tumor appears beefy red from rich vascularity; multiple cysts and cavernous spaces may be seen in a cross-section; in areas, the tumor may appear yellow from lipid deposition.
Microscopic Features
Histologically the tumor is composed of three groups of cells: 1.endothelial cells, 2. pericytes, and 3. stromal cells. Whether all three sets of cells participate in the neoplastic process has been the subject of debate; also unresolved is the question of whether these cells interconvert. The cardinal feature of the tumor is the presence of numerous capillary channels that form an anastamosing plexiform pattern, and are, lined by a single layer of plump endothelial cells. The capillary channels are surrounded by reticulin fibers, best demonstrated by reticulin stains. The pericytes are difficult to discern with light microscopy and are best visualized with electron microscopy. They lie just outside the periendothelial basement membrane and are themselves completely surrounded by a basement membrane. Between the capillary structures are numerous polygonal cells, the interstitial cells, or stromal cells, with foamy clear cytoplasm. The stromal cells are generally laden with lipid.
Intense research has centered around the question of the origin and nature of the stromal cells, ever since Lindau gave an accurate description of the histology of these tumors in 1926. Over the years, numerous investigators have tried to solve the problem by utilizing various newly developed histologic techniques. In each case, the hypothesis proposed on the basis of the new technique was later shown on the basis of a yet newer technique to be invalid. Several hypotheses are summarized in Table-2.
TABLE -2
Hypotheses on the Origin and Nature of Stromal Cells in Hemangioblastoma
Author and Year
Technique Used
Findings
Interpretations/Conclusions
Lindau (1926)
Light microscopy
Stromal cells are often filled with lipids.
Disturbance of circulation in tumor tissue is accompanied by stagnation of lymph rich in lipids which are phagocytosed by the endothelial tumor cells.
Various neuropathologists ( 1930-1940)
Metallic stains for neuroglia and microglia
Stromal cells are not stained with metallic stains.
Stromal cells are not derived from glial cells
Cancilla and Zimmerman (1965)
Electron microscopy
Close ultrastructural similarity between endothelial cells and stromal cells
Hemangioblastomas are composed of a single cell type which originates from the endothelium.
Castaigne et al.(1968)
Electron microscopy
Ultrastructural observations
Stromal cells are neoplastic reticulum cells: endothelial cells and pericytes are non-neoplastic
Cervos-Navarro (1971) and Kawamura et al. (1973 )
Electron microscopy
Overlapping ultrastructural features between endothelial cells, pericytes, and stromal cells
Stromal cells are derived from "vasoformative elements" (endothelium and pericytes).
Jellinger and Denk (1974)
Red-cell adherence for blood group isoantigens
Endothelial cells in tumor contain blood group isoantigens; stroma cells do not.
Stromal cells are unlikely to be of endothelial origin
Spence and Rubinstein (1975)
Organ culture and electron microsocopy
Endothelial cells, pericytes. and stromal cells maintain their identity in organ culture.
Endothelial cells. pericytes, and stromal cells are all neoplastic and replicate in parallel with one another. Interconversion between these cells does not occur.
Jakobiec et al. (1976)
Electron microscopy and lipid analysis
Lipid in stromal cells is mostly cholesterol stearate, a plasma lipid; fibrous astrocytes are in different stages of lipidization
Stromal cells represent lipidized astrocytes; source of lipid is blood plasma; lipidization alters tinctorial properties so that cells no longer stain with metallic stains: occurrence of hemangioblastomas exclusively in nervous system further attests to the neuroectodermal origin of tumor, as opposed to origin from angiogenic mesenchymal elements.
Kepes et al. (1979)
Immunoperoxidase method for detection of glial fibrillary acidic protein (GFAP)
In about half the cases, stromal cells are negative for GFAP; in the remainder, variable amounts of GFAP are demonstrated.
Stromal cells may be of heterogeneous origin: deposition of lipid droplets in cells of diverse origin may make them appear similar.
Jurco et al. (1982)
Immunoperoxidase method for factor VIII-related antigen (VIII R:Ag) and GFAP
All cases show positive staining for VIII R:Ag in stromal cells; astrocytes staining positively for GFAP are noted peripherally and centrally in the tumor, but all stromal cells are negative for GFAP
Stromal cells are of endothelial origin: occasional stromal cells identified by other investigators as reacting positively for GFAP may represent stromal cells capable of ingesting extracellular GFAP derived from reactive astrocytes within tumor or may be lipidized astrocytes.
Tanimura et al. (1984 )
Immunoperoxidase technique for GFAP, S-I00, and factor VIII-related antigen
Most stromal cells are GFAP-positive: a variable number scattered throughout the tumor are positive for S-I00. Factor VIII-related antigen staining is negative except in endothelial cells lining the capillaries.
Stromal cells consist of heterogeneous cell populations
Holt et al. (1986)
Immunohistochemistry with GFAP, factor VIII-related antigen, and UEAI-a sensitive marker of endothelial cells
Factor VIII-related antigen and UEAI are limited to endothelial cells, and GFAP is present only in trapped astrocytes
No convincing evidence that stromal cells are derived from endothelial. pericytic, or astrocytic cells: origin remains uncertain.
Kamitani et al. (1987)
Light and electron microscopy
Stromal cells demonstrate pericytic and leiomyoblastic characteristics
Stromal cells are closely related to pericytes and smooth muscle cells, supporting the concept that pericytes serve as precursors to stromal cells.
Frank et al. (1989)
Immunohistochemistry with 17 different cell-type specific markers
No consistent staining of stromal cells for markers for endothelial, epithelial, chromaffin, or smooth muscle origin.
Stromal cells of cerebellar hemangioblastoma are not endothelial, neural. epithelial, pericytic, or neuroendocrine in origin and probably are of undifferentiated mesenchymal origin
Morii et al. (1993)
In situ hybridization, Northern blotting, PCR analysis
Vascular endothelial growth factor (VEGF) mRNA highly expressed in the stromal cells but not in the endothelial cells
VEGF secreted from stromal cells plays an important role in endothelial cell proliferation in hemangioblastomas.
Diagnostic Studies
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) is the technique of choice to visualize hemangioblastomas because it is highly sensitive and provides better anatomic definition than computed tomography (CT). Further, the multiplanar capabilities of MRI are ideally suited to provide maximal anatomic definition of these lesions. An additional advantage to MRI is that it is better suited for imaging lesions in the posterior fossa where hemangioblastomas have a predilection to occur. Cerebellar and supratentorial hemangioblastomas typically demonstrate a sharply marginated smooth cyst border and an enhancing tumor nodule. Prior to the administration of a gadolinium-containing contrast material, the mural module is hypointense to isointense on T1-weighted and is hyperintense on T2-weighted images. Administration of an MR contrast agent typically demonstrates intense enhancement of the mural nodule. Non-cystic hemangioblastomas enhance uniformly following the administration of the contrast agent. Spinal cord hemangioblastomas may be characterized by an intramedullary vascular nodule, enlarged draining veins, diffuse enlargement of the cord, or an intramedullary cyst.
Computed Tomography
On computed tomography (CT), hemangioblastomas typically enhance intensely after the injection of a contrast agent, with either a homogeneous or a mottled appearance; in some cases there may be a high-density rim with central lucency from an intratumoral cyst. Dilated vessels may be seen in the vicinity of the tumor. The cystic tumors appear as sharply-defined, low-density lesions with or without a mural nodule. The attenuation values of the cyst may be the same as or slightly higher than those of cerebrospinal fluid. Bony artefact may be problematic in imaging the posterior fossa. The mural nodule, if present, enhances intensely on contrast injection, but the cyst margin itself generally does not enhance. If a cyst is seen alone, without a nodule, in a patient with known VHL complex, one has to conclude that the nodule is too small to be defined by CT. In such instances, angiography is mandatory to locate the mural nodule, because the cyst is bound to recur unless the mural nodule is removed. Typically, solid tumors are uniform, isodense or hyperdense, and enhance with contrast. Important CT characteristics include marked contrast enhancement of the nodule, proximity of the mural nodule to the pial surface, large size of the cystic component of the tumor compared with the mural nodule, and isodensity of the nodule on preinfusion CT.
Conventional Angiography
On vertebral angiography, four different vascular patterns may be observed: (1) a vascular mural nodule within an avascular cyst; (2) a doughnut ring of abnormal vessels surrounding an avascular space representing an intratumoral cyst; (3) a large, solid vascular mass; and (4) multiple small, widely separated vascular nodules. Since vertebral angiography is highly sensitive in detecting small tumor nodules, it is especially helpful in detecting high spinal lesions as well; thus the neck should be routinely imaged during vertebral angiography in patients with the VHL complex. A solid hemangioblastoma in the anterior surface of the cerebellum abutting against the petrous bone may on occasion simulate a cerebellopontine angle tumor on CT; in such instances the characteristic angiographic pattern will help in their differentiation. Superselective angiography has been advocated by several authors for solid hemangioblastomas of the fourth ventricle and medulla. Further, interventional techniques have proven useful in devascularizing hemangioblastomas with deep large arterial feeding vessels. Some authorities have reported the use of preoperative embolization as a useful technique to devascularize the lesion and facilitate the operative extirpation.
Ultrasonography
Intraoperative Doppler ultrasound has been a useful adjunct in the surgical treatment of hemangioblastomas. The majority of lesions are easily detected with standard gray scale ultrasound. However, color Doppler flow images provide improved delineation of lesions compared with the images produced with standard gray scale ultrasound. In addition, some lesions are visible only with color Doppler imaging because they are isoechoic to the spinal cord on gray scale ultrasound.
Hemangioblastomas in Other Locations
Supratentorial Hemangioblastomas
Supratentorial hemangioblastomas are distinctly rare. Sporadic case reports have appeared in the literature. These tumors may occur in the brain parenchyma of the frontal, parietal, temporal, or occipital lobes, in the corpus callosum or basal ganglia, along the walls of the lateral and third ventricles, in the choroid plexus, and in the leptomeninges. Clinical features vary widely, depending on the location of the tumor. Association with other features of von Hippel-Lindau complex has been demonstrated in 10 of 63 reported cases. In 7 of the 63 cases, polycythemia was present. Twothirds of the tumors have been solid, with the remainder having at least some cystic component. A congenital cystic supratentorial hemangioblastoma in a 3-week-old infant was reported. Although, the radiologic features of supratentorial lesions may closely resemble those of hemangioblastoma in the cerebellum, hemangioblastoma is seldom entertained in the differential diagnosis, because of its rarity in the supratentorial compartment, unless the patient has overt signs of the von Hippel-Lindau complex.
Hemangioblastomas Involving the Optic Nerve
Optic nerve hemangioblastomas are very unusual, with fewer than a dozen documented cases. They must be differentiated from retinal angioma, a much more common lesion. Optic nerve hemangioblastomas typically present with progressive visual deficits with or without headache. As retinal angiomas can also cause progressive visual loss, it is important to correlate the degree of visual loss with the extent of retinal disease evident on funduscopic examination. Visual loss out of proportion to the degree of retinal disease should prompt an evaluation for an optic nerve hemangioblastoma. The lesions are typically prechiasmatic and are histologically indistinguishable from hemangioblastomas found in other locations.
Hemangioblastomas of the Spinal Cord
Hemangioblastomas of the spinal cord represent 1.5 to 2.5 percent of all spinal cord neoplasms. The median age of onset of symptoms is around 35 years. In patients with the VHL complex, the spinal symptoms may appear concurrently with or (more often) after the onset of cerebellar or retinal symptoms. Stated differently, the onset of spinal symptoms in a patient with known VHL complex should alert the clinician to the possibility of a spinal hemangioblastoma. Sixty percent of hemangioblastomas are intramedullary and are located in the dorsal half of the spinal cord near the midline. Extramedullary intradural hemangioblastomas tend to be attached to the posterior nerve roots. The thoracic spinal cord is most frequently involved, with the cervical segments next in frequency. Syringomyelia is associated with more than half the cases of spinal hemangioblastoma. The development of syringomyelia is analogous to the development of a cyst in a cerebellar hemangioblastoma, i.e., it arises from transudation of fluid from the tumor capillaries and tubular dissection along the gray matter near the central canal, which offers the least resistance. Syringomyelia in patients with the VHL complex has never been reported without the accompaniment of a hemangioblastoma in the spinal cord except in the cervical region, where it may be related to a similar tumor in the brain stem. The clinical presentation is variable. Three common types are described: (I) slow evolution of long-tract symptoms and signs (posterior column and corticospinal tracts), with or without radicular symptoms; (2) subarachnoid hemorrhage without focal neurological signs; and (3) subarachnoid hemorrhage with abrupt onset of long-tract signs. Plain roentgenograms of the spine show erosion of the pedicles and widening of the spinal canal in about half of the cases. The myelographic appearance is indicative of an intramedullary mass, and. in some cases, serpentine filling defects may simulate an arteriovenous malformation (in the latter, the spinal cord is not usually enlarged). Whereas CT may demonstrate only cord widening, MRI precisely defines the anatomy of the lesion. Spinal angiography is most helpful in arriving at the specific diagnosis. A densely staining tumor nodule is visualized, with evidence of rapid circulation through the tumor. Preoperative embolization of feeding vessels may be considered at the completion of angiography. With the use of the operating microscope and bipolar coagulation, total excision of the tumor is possible in most cases. The dura should be opened carefully to avoid damage to the dilated pial vessels. The tumor should be dissected in the plane between the tumor and the parenchyma of the spinal cord, with meticulous coagulation of all feeding vessels. No attempt should be made to biopsy the tumor or excise it piecemeal.
Hemangioblastomas of the Peripheral Nerves
The only reported occurrences of hemangioblastoma outside the central nervous system are two individual case reports of peripheral nerve hemangioblastomas. These are obviously very rare lesions, In both reported cases, the patient presented with pain and loss of function of a single peripheral nerve; both cases responded well to surgical treatment. These lesions were radiographically and pathologically identical to those within the central nervous system.
Angiomatosis Retinae: von Hippel's Disease
The lesions of angiomatosis retinae start as small, discrete foci of dilated capillaries during childhood or adolescence. With time, the angioma appears as a raised or globular reddish mass fed by dilated, tortuous arterioles and drained by engorged serpentine veins, which result from low vascular resistance in the tumor. Although most retinal angiomas become evident in the second and third decades, the age of onset is highly variable. More than 70 percent of patients with the VHL syndrome develop retinal angiomatosis by age 60. Fluorescein angiography confirms hyperdynamic circulation with intense staining of the nodule and early venous return. Although most commonly located in the peripheral parts of the retina, retinal hemangioblastoma has been observed in the macula, at the border of the optic disc, or on the optic disc. These tumors generally affect both eyes. In longstanding cases, a gray-white exudate of variable degree surrounds the mass, the feeding artery, and the draining vein. The exudation is thought to result from increased capillary permeability due to the presence of fenestrations in the lining endothelium. Frequently there is retinal separation, massive gliosis and retinal edema. On occasion, a macular star or Papilledema develops. Recurrent bleeding from the lesion is not infrequent. In late stages, secondary glaucoma with narrowing of the anterior chamber, dilation of the pupil and extreme congestion may render the eye functionally useless. In uncomplicated cases of hemangiomas situated in the peripheral retina, the visual symptoms are minimal or absent in the early stages. At this stage, the lesion may be diagnosed by diligent funduscopic examination, especially in patients with a known family history of this condition. Scotomas or a decrease in visual acuity results when there are complicating factors such as exudation, retinal separation, gliosis or hemorrhage; visual loss may be disproportionately severe when the lesions are located in the macula or on the optic disc. The histologic appearance of the retinal lesions is similar to that of hemangioblastomas occurring in the cerebellum or spinal cord. Hemangioblastoma of the retina should be distinguished clinically from arteriovenous malformations of the retina, Coat's disease, glioma of the retina, melanoma of the choroid, Eales' disease and multiple retinal aneurysms. Photocoagulation is the treatment of choice because it is highly effective for small lesions and carries very little morbidity. The safety and efficacy of treatment for small lesions underscores the importance of early diagnosis of retinal angiomas. Despite successful treatment, however, new lesions may develop in other parts of the retina and become apparent with continued follow-up examinations.
Related Conditions
Until recently, cerebellar hemangioblastomas were the most common cause of death in the VHL syndrome. With improved methods of surgical treatment of posterior fossa lesions, the optimal management of extracranial related conditions becomes all the more important. Abdominal manifestations of the VHL syndrome are often asymptomatic but are associated with considerable morbidity and mortality. The identification of genetic probes that flank the VHL gene has made it possible to detect individuals who have inherited the VHL trait in families that are at risk. Focused screening programs directed to such individuals will make possible presymptomatic diagnosis of these lesions and thereby reduce their overall morbidity and mortality.
Renal Cell Carcinoma
Renal lesions are characterized as either (I) benign cysts or (2) solid renal carcinomas with or without cystic degeneration. Several authors have demonstrated that renal cysts form a histopathologic continuum from benign cysts to malignant cysts that harbour foci of renal cell carcinoma in their walls. Unlike sporadic benign renal cysts found in the general population, the walls of cysts in patients with the VHL complex often have foci of occult renal cell carcinoma. Renal cell carcinoma is found in one-fourth of patients with the VHL complex. The frequency tends to be higher in autopsy series than in clinical series because these neoplasms tend to remain silent in the early stages. The tumor may be the last one to be detected unless CT or MRI of the abdomen is done as a screening procedure in patients and relatives at risk. As more patients survive the common and potentially fatal posterior fossa hemangioblastoma, more cases of renal cell carcinoma will be observed in the respective kindreds. The renal cancer differs from its sporadic counterpart in its earlier age of onset, slight male predominance, multicentricity and synchronous or metachronous bilateral involvement. These factors pose special problems in surgical therapy. Some have suggested bilateral nephrectomy, renal dialysis for 5 years, and a renal transplant if the patient survives that long without recurrence of tumor. However, the annual attrition rate of 10 to 12 percent for patients on hemodialysis or after transplantation remains a distressing concern. In addition, a 4 to 6 percent incidence of de novo neoplasia and an incidence of up to 38 percent of recurrent or metastatic carcinoma have been reported in transplant recipients, presumably as a result of immunosuppression. One may expect that these incidence rates will be even higher in patients with the VHL complex, who are genetically predisposed to neoplasia. Therefore, conservative extirpative surgery with preservation of as much functional renal tissue as possible appears justified in these patients. Cytogenetic evaluation has revealed that the chromosomal aberrations in renal cell carcinomas associated with the VHL complex are similar to those in spontaneous renal cell carcinomas and result from a deletion in the short arm of chromosome 3 (3p 11). Furthermore, the fact that a deletion in 3p was noted in renal cell carcinomas in different stages of development suggests that a rearrangement of the 3p region is the first genetic change during oncogenesis. The genetic aberrations most frequently detected were a chromosome exchange between 3p and 5q (by which genetic deletions from chromosome 3 were added to chromosome 5), a 3p deletion, and a non-disjunctional loss of an entire chromosome 3. These findings are in keeping with a current hypothesis that holds that the central event in oncogenesis for these lesions is the loss of function of a tumor suppressor gene on the short arm of chromosome 3.
Pheochromocytoma
Pheochromocytoma is seen in about 10 percent of patients with the VHL complex, whereas about 23 percent of patients diagnosed initially as having pheochromocytoma will have either the VHL complex or the syndrome of multiple endocrine neoplasia (MEN). A review of reported families with the VHL complex suggests that some families are more prone to the development of this tumor than others. The tumor tends to occur bilaterally, implying that persons with the VHL complex with unilateral pheochromocytoma are at risk of developing a second tumor in the opposite adrenal. As a corollary, a patient who presents with bilateral pheochromocytomas should be examined for other evidence of heritable syndromes in which bilateral pheochromocytomas are known to occur: (I) von Recklinghausen's neurofibromatosis, (2) the VHL complex, (3) multiple endocrine neoplasia syndrome, subtype IIA, and (4) simple familial pheochromocytoma. In certain instances, adrenal medullary hyperplasia may precede the actual development of tumor. Familial pheochromocytomas occur at a younger age and are more frequently multifocal than their sporadic counterparts. Pheochromocytomas were so named because of their affinity for chromium salts. As with tumors arising from the anterior pituitary gland, there is imperfect correlation between the functional activity of the tumor and its tinctorial properties with light microscopy. All catecholamine-secreting tumors arising from the adrenal medulla are now designated as pheochromocytomas regardless of whether they stain with chromium salts. Thus, the older term nonchromaffin paraganglioma for a functioning tumor of the adrenal medulla that fails to stain with chromium salts is unacceptable. Ten percent of pheochromocytomas are malignant, but the malignant variant cannot be distinguished from the benign tumor on histologic grounds; the sole criterion is the development of metastases, with the malignant tumor appearing at sites where chromaffin tissue does not normally occur. The cardinal symptoms are episodic headaches, excessive sweating, palpitation, nervousness, and tremor. Headaches in a patient with the VHL complex may be due to a cerebellar tumor with increased intracranial pressure, to catecholamine excess from a pheochromocytoma, or to both. In the first case, the headaches tend to be suboccipital in location, dull, persistent, and often associated with nausea and vomiting. The headaches from catecholamine excess tend to be paroxysmal, pounding or throbbing in character, and to last from a few minutes to several hours; they tend to be associated with other paroxysmal symptoms such as feelings of apprehension, excessive truncal sweating, tremors, tachycardia, palpitations and anginal pain. There may be paroxysmal or nonparoxysmal elevations in blood pressure. Occasionally the first manifestation of pheochromocytoma is a dangerous elevation of blood pressure noted during an elective operative procedure in a patient with the VHL complex. Impaired glucose tolerance and hypermetabolism with weight loss are often observed. The best screening test is a urinary metanephrine assay in a single voided (spot) specimen. Values ranging from 1.0 to 2.2 µg of metanephrine per milligram of creatinine are considered suspicious, and 24-h urine specimens should be examined for metanephrine, vanillylmandelic acid (VMA), and catecholamines. Plasma catecholamine levels are more expensive and technically more difficult to measure and are indeed less reliable because of wide swings in their values even under physiologic conditions. The tumor is best localized by abdominal MRI. Therapy is directed toward resection of the tumor(s) with intensive monitoring and pharmacologic control of blood pressure perioperatively. Ninety percent of these tumors are surgically curable.
Polycythemia
Hemangioblastoma is the only central nervous system tumor to be associated with polycythemia. Polycythemia has been reported in 9 to 20 percent of cases of posterior fossa and supratentorial hemangioblastomas but has not been reported with purely spinal lesions. There is erythrocytosis with no associated splenomegaly or increase in the white cell or platelet count. The red cell volume increases, but the plasma volume remains normal. The red cell life span is within the normal range. There is no evidence of accelerated red cell destruction in the liver or spleen. The rate of red cell synthesis is increased, and the red cell iron turnover is increased proportionately. The bone marrow may show erythroid hyperplasia. Erythrocytosis in patients with hemangioblastoma is believed to be due to the unregulated secretion of erythropoietin or an erythropoietin-like substance by the neoplastic tissue. Histologic sections of a hemangioblastoma may show areas of hematopoietic activity, but this is insufficient to induce polycythemia. Injection of tumor cyst f1uid or extracts from solid tumors has induced erythrocytosis in experimental animals. Erythropoietin is known to promote the differentiation. proliferation, and maturation of red cell precursors in the bone marrow. It is a glycoprotein with a molecular weight of approximately 40,000; it migrates as an α-globulin during electrophoresis. Although the kidneys are thought to produce erythropoietin under physiologic conditions, the exact role the kidneys play in its production remains controversial, inasmuch as it has not been possible to isolate erythropoietin from kidney extracts. Two alternative roles for the kidneys have been put forward. They may produce an enzyme (erythrogenin) which may act on a substrate in the plasma (erythropoietinogen) which is produced by the liver; or they may synthesize erythropoietin in an active form that may be rapidly inhibited by a complex lipid. The stimulus for secretion of erythropoietin is tissue hypoxia. Erythropoietin levels are increased in patients with anaemia, renal artery stenosis, renal cysts, and renal neoplasms. Thus, erythrocytosis in a patient with the VHL complex may be due to a renal lesion or to a nervous system hemangioblastoma. The erythrocytosis induced by hemangioblastoma may improve after total excision of the tumor or irradiation, only to reappear with recurrence of the tumor. Ultrastructural studies of certain hemangioblastomas have shown granules thought to represent intracellular erythropoietin, but positive proof of their identity is lacking. Table-3 gives an outline of the diagnostic workup of a patient with suspected VHL complex
TABLE-3
Diagnostic Workup of a Patient with Suspected VHL Complex
1.
Elicitation of family history and clinical examination
2.
Detailed funduscopic examination by an ophthalmologist and possible fluorescein angiography if a lesion is found; fundus photographs for follow-up evaluation; visual acuity and fields
3.
Hematocrit and red blood cell count
4.
Magnetic resonance imaging in axial, sagittal, and coronal planes without and with contrast enhancement; upper cervical spine area included
5.
Arteriography with subtraction technique; upper cervical area included
6.
Urine for metanephrine screen; if positive, 24-h VMA and catecholamine determinations
7.
Abdominal CT/MRI without and with contrast enhancement; special emphasis on pancreatic, renal, and suprarenal areas
8.
Spinal angiography if a spinal lesion is suspected or detected on MRI
Treatment
Surgery of Posterior Fossa Hemangioblastoma
The operative exposure is through a standard posterior midline or paramedian approach for vermis or cerebellar hemispheric lesions, respectively. A transoccipital transtentorial approach may be desirable for lesions located in the anteroventral surface of the cerebellum abutting against the petrous bone. The patient may be in the sitting, semi sitting, prone, or semiprone position, depending on the surgeon's preference. Magnification and bipolar coagulation are mandatory. If the dura is extremely tense from a cerebellar cyst, the cyst may be tapped through the dura or through a small durotomy, taking care to avoid striking the mural nodule. The location of the tumor is marked on the surface by dilated pial vessels. The mural nodule is quite small and generally is located close to the pial surface. It suffices to excise the mural nodule and drain the cyst; it is not necessary to excise the cyst wall, since it is not composed of tumor. However, drainage of the cyst alone without removal of the mural nodule invites recurrence of the cyst in the immediate postoperative period. Solid tumors should be dealt with in the same way as vascular malformations. No attempt should be made to biopsy the tumor or to remove it piecemeal. One should recall the histologic appearance of the tumor, composed of a meshwork of vascular spaces lined by endothelial cells but with no contractile elements. Thus, an ill-conceived biopsy may lead to profuse, uncontrollable bleeding, and piecemeal removal of the tumor invites disaster. The surgeon should cautiously make a cortical incision until the surface of the tumor is reached; at this point the white matter adjacent to the tumor should be dissected all around, coagulating the feeding vessels through the dissection. If the bipolar coagulator is used in the cutting rather than coagulation mode, there is minimal charring at the forceps tips and less stickiness, allowing more rapid dissection around the tumor. The tumor should be excised in toto as a single mass. It is worth noting that solid tumors tend to involve the brain stem more often than cystic ones. Tumors arising primarily from the brain stem should be closely scrutinized. Those with attachment to the floor of the fourth ventricle in the midline are hazardous to remove because of cardiorespiratory complications. Those attached laterally to the inferior cerebellar peduncle may be removed with surprisingly slight morbidity. Multiple hemangioblastomas in the posterior fossa pose a special and difficult problem. If the tumors are at least 0.8 to 1 cm in diameter and are easily accessible, they may be removed in the same way as solitary tumors; but if they are smaller and are deep, they may be difficult to find. This is especially true if a prior posterior fossa exploration has led to adhesions and distorted anatomy. In such situations, it may be prudent to follow these tumors on serial MRI scans and to expose them when they are just large enough to be detected, or better still, when they develop a cyst. With further refinements in CT-assisted and MRI-assisted stereotactic approaches to deep brain lesions, it is now possible to approach them with greater certainty and precision and to eradicate them. In all instances in which the posterior cranial fossa is explored for hemangioblastoma, the dura should be closed in a watertight manner, using grafts if necessary. Given that recurrences of these tumors are common and that the tumor is known to be multicentric, the surgeon should be prepared for re-exploration of the posterior fossa. Re-exploration becomes extremely difficult if there are adhesions due to prior failure to close the dura adequately.
Radiation Therapy
Reliable data on the effects of radiation in hemangioblastomas based on prospective controlled studies on a large series of patients are not available. Recent evidence, however, suggests that high-dose irradiation (4500 to 5000 rad given over 4.5 to 5 weeks) may significantly reduce the size of the tumor or at least retard its rate of growth; decrease its vascularity; and extend the symptomfree interval and survival time of patients. One tangible effect of radiation on the tumor, besides the decrease in size and vascularity seen on angiograms, is the resolution of erythrocytosis. However, it should be emphasized that, unlike surgical resection, radiation treatment is not curative. There is little justification for considering radiation therapy for this benign lesion in the cerebellum, spinal cord, or cerebral hemisphere. For inoperable lesions in the brain stem or for solid lesions of the cerebellum that extend into the brain stem via the cerebellar peduncles, however, radiation therapy may be the only choice. Several recent reports have advocated stereotactic radiosurgery for recurrent and multifocal hemangioblastoma that involves the brain stem. Stereotactic radiosurgery offers added benefits over conventional radiation for patients with the VHL complex who are at risk of developing further lesions with time. Because the radiation is tightly focused, subsequent lesions can be treated without significantly increasing the radiation exposure of the surrounding normal brain.
Prognosis
The long-term prognosis is best for a solitary cystic tumor in the cerebellum. Given that 70 percent of cerebellar hemangioblastomas are cystic and the tumors are always benign, most non-familial hemangioblastomas can be resected with minimal morbidity and less than 2 percent mortality. The solid tumors tend to be larger and more vascular and tend to involve the brain stem; for them, the operative mortality is on the order of I5 percent. Lesions with deep midline attachment to the medulla oblongata are invariably lethal. Occurrence of multicentric malignant genitourinary and other visceral tumors in patients with the VHL complex becomes a major determinant in assessing expected survival even after successful removal of a cerebellar hemangioblastoma. The multicentric origin of these tumors within the neuraxis in patients with the VHL complex adds further to the morbidity and mortality; the recurrence rate is 3 to 10 percent after total excision of the initial tumor, and the symptom-free interval averages 5 years.
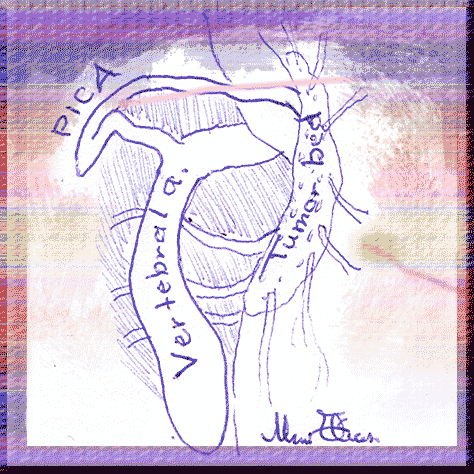
A young woman, how underwent first surgery one year ago for as be diagnosed incorrectly, arachnoid cyst was seen with deterioration of her clinical picture with ataxia and left sided weakness and was sent to MRI, which showed enlargement of the cystic mass with descendance of the tonsils down to C2. Hemangioblastoma was suspected and she was operated. As you figure in the video and graphic presentation, the hemangioblastoma was surrounding the left vertebral artery and the origin of the PICA and having several hundreds of small feeders from the lateral medulla. The difficult situation in the case was the complete penetrance of the vertebral artery in the mass, which hold suspicion of aneurysm all the time of the lengthy operation. The mass was removed at last in toto and the patient postoperative period was uneventful.