|
Amyotrophic lateral sclerosis
(ALS) or “Lou Gehrig’s disease” (after the
famous American baseball player who was diagnosed with
the disorder) is a rare degenerative disorder of motor
neurons of the cerebral cortex, brain stem, and spinal
cord that results in progressive wasting and paralysis
of voluntary muscles. The age of onset is in middle
adult life. It progresses rapidly and most of the
affected individuals die within 3–5 years from onset of
symptoms.
The cause of the disease is
still elusive, except in familial cases (about 10%)
where the most common cause is linked to mutations in
the gene encoding cytosolic copper–zinc superoxide
dismutase (SOD1, an enzyme responsible for scavenging
free radicals).
Essential features of ALS are progressive signs and
symptoms of lower motor neuron dysfunction. These
include focal and multifocal weakness, atrophy, cramps,
and fasciculations associated with corticospinal tract
signs (spasticity, enhanced, and pathological reflexes)
in the absence of sensory findings. Usually, the
symptoms initially affect a limb, but in some cases
there might be a bulbar onset with difficulties in
speaking and swallowing. Regardless of the part of the
body first affected by the disease, muscle weakness and
atrophy spread to other parts of the body as the disease
progresses.
No treatment prevents, halts, or
reverses the disease, although marginal delay in
mortality has been noted with the drug riluzole.
ALS is a difficult disease to
diagnose as there are many other, more treatable
diseases which mimic it. Thus, the diagnosis is
primarily based on the symptoms and signs observed by
the physician and a series of tests to rule out other
diseases. To be diagnosed with ALS, patients must have
signs and symptoms of both upper and lower motor neuron
damage that cannot be attributed to other causes.
Electromyography (EMG) provides objective evidence of
lower motor neuron involvement. When the lower motor
neuron involvement is severe, the upper motor neuron
signs may be masked and its involvement can be missed.
In this context, MR methods can be very useful to detect
early involvement of upper motor neuron involvement,
potentially shortening the time to diagnosis.
Recent MRI studies have shown the diagnostic utility of
hyperintensity of the corticospinal tract on FLAIR
sequences in ALS. Other studies, using diffusion images
or quantitative morphometry, have provided evidence of
abnormalities of extramotor areas, supporting the view
that ALS is a multisystem degenerative disease.
1H-MRS
Among the recent MR technologies used
to improve detection of upper motor neuron involvement,
1H-MRS can provide insights into the metabolic integrity
of these pathways. Not surprisingly, in the brain of ALS
patients, NAA is decreased in a spatially dependent
manner that reflects its pathologic distribution and mI
is often increased in the motor cortex. This has led to
the proposal of using the NAA/mI ratio, (Fig-1 and 2)
which would enhance the ability of MRS to distinguish
patients with ALS from control subjects. This might
become a useful biomarker in ALS. In recent pilot
studies, 1H-MRS has been used to monitor whether
riluzole does have a mild disease-altering effect. The
finding of an increase in NAA/Cr ratio in the motor
cortex of ALS patients after a brief treatment with
riluzole should be confirmed in controlled studies
involving larger patient populations.
Since the main goal is to study the regional
distribution of metabolites in the corticospinal tract,
both single-voxel 1H-MRS and 1H-MRSI can be used. The
assessment of both mI and NAA should be pursued,
therefore short TE (30–35 ms) acquisitions are
preferable.
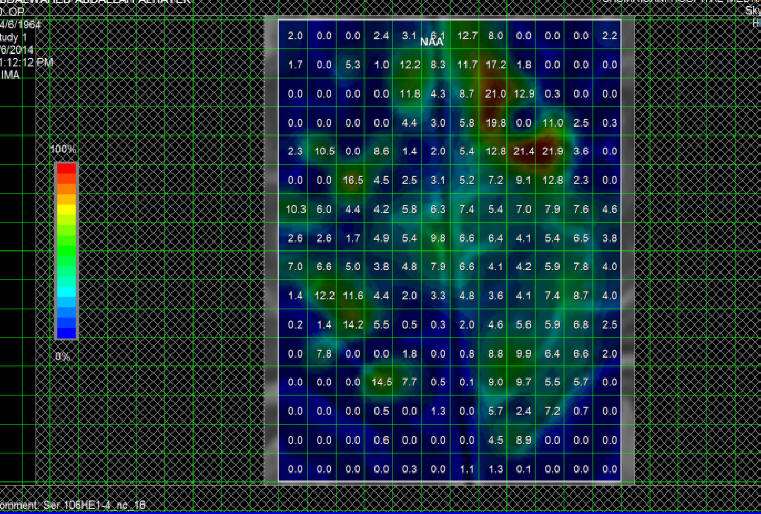
Fig-1: NAA distribution over the motor and premotor
cortex in patient with ALS.
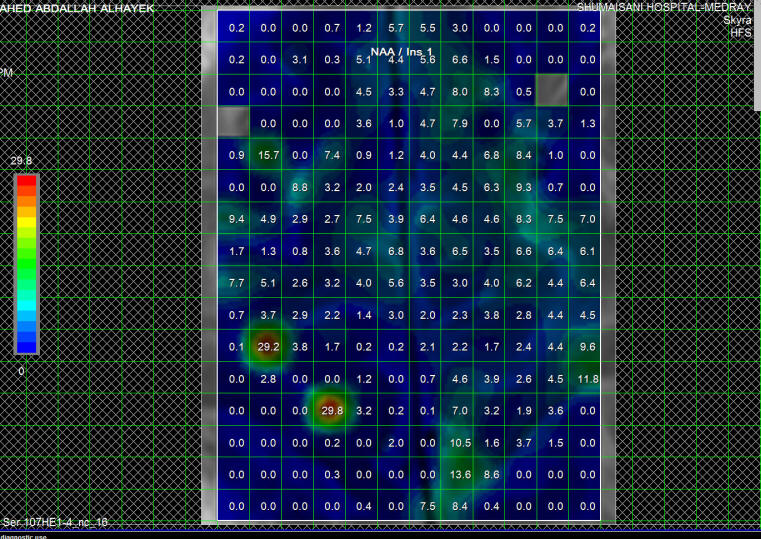
Fig-2: Same patient as in fig-1 with NAA/MI ratio in the
same area.
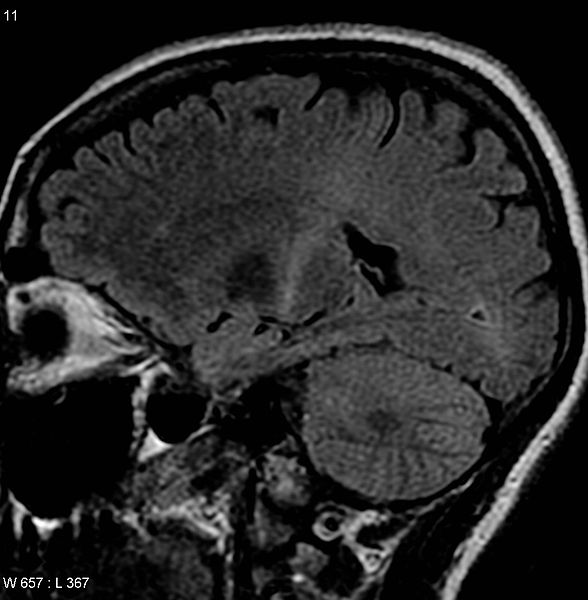
Figure-3: This MRI (parasagittal FLAIR) demonstrates
increased T2 signal within the posterior part of the
internal capsule and can be tracked to the subcortical
white matter of the motor cortex, outlining the
corticospinal tract, consistent with the clinical
diagnosis of ALS. However, typically MRI imaging is
unremarkable in a patient with ALS.
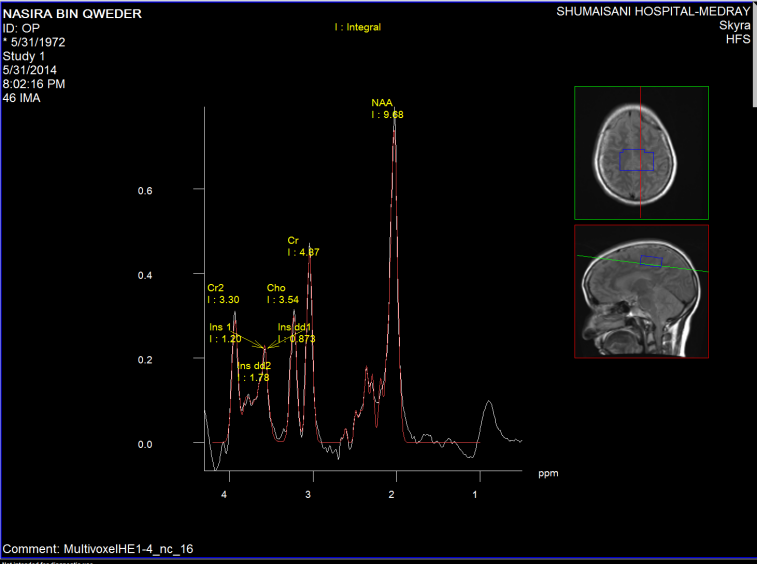
Figure-4: PCD C3-4 with spinal cord compression with
spinal artery syndrome can mimic the clinical picture of
the patient. This patient suffering from muscle atrophy
for 16 years with inability to walk with full blown
picture of ALS. The high levels of NAA is against the
ALS in MRS.
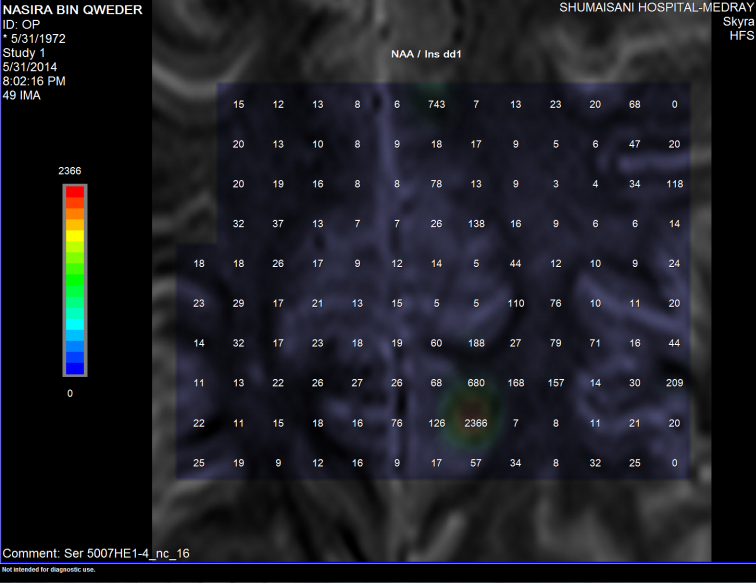
Figure-5: The NAA/Inositol ration is not decreased,
instead it is increased, ruling out the presence of ALS.
Figure-6: MRI of patient with ALS with the increased
signal of T2 in the posterior part of the internal
capsule left side.
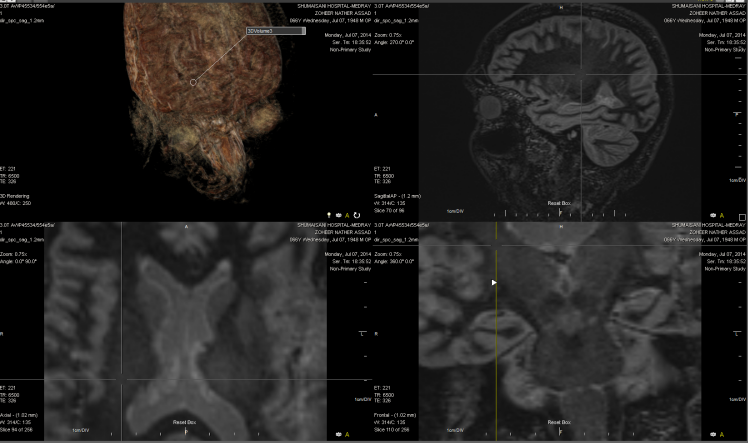
Figure-7: The same signal in the right side of the same
case of figure 6.
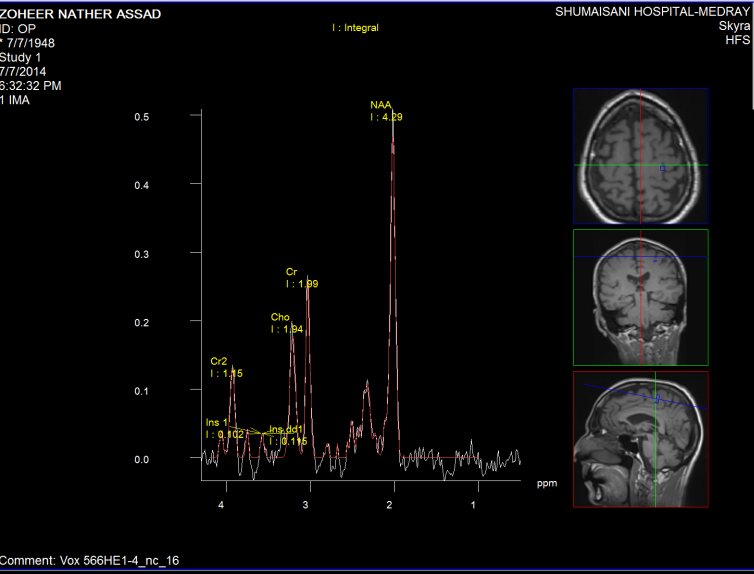
Figure-8: The same patient in figure-6 with normal
pattern of Short TE spectroscopy.
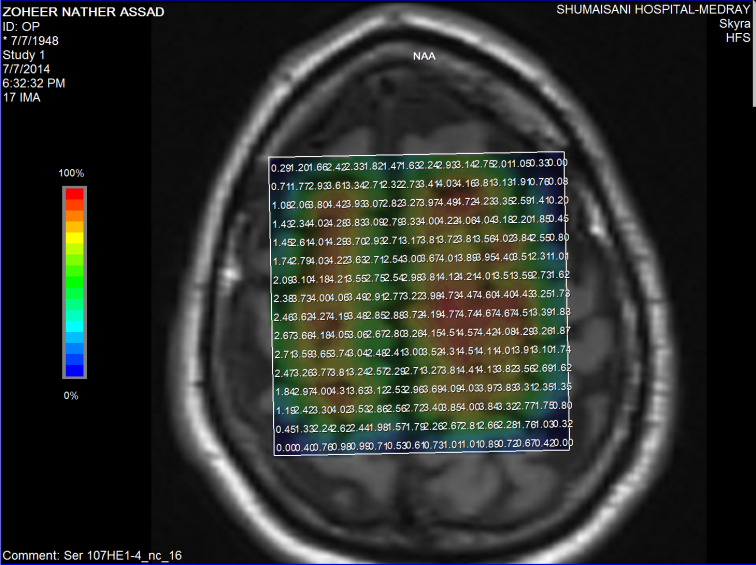
Fifure-9: The same patient with normal values of NAA.
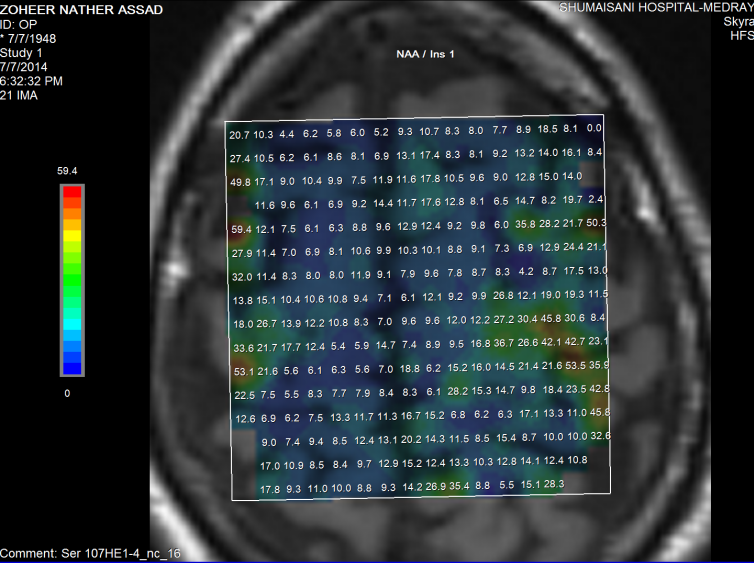
Figure-10: The same patient in figure 6 showing normal
ratio of NAA/Inositol. From these data from figure 6-10,
it seems that the presence of T2 shadows in the
posterior limb of the internal capsules are more
reliable than the spectroscopic data.
|